

导语:不明原因的反复鼻衄,这种罕见病因不能忽视!
临床中遇到不明原因的反复鼻衄,伴肝脾肿大,且血常规提示全血细胞减少,会考虑
什么疾病呢?
有些疾病虽然罕见,却对患者的生活产生了深远影响。来自南方医科大学南方医院的
14 岁青少年男性患者面临着反复鼻衄的困扰,经过骨髓检查和实验室酶学检查,细心
敏锐的医生用葡萄糖脑苷脂酶活性检测揭开该罕见病的神秘面纱。
不明原因的反复鼻衄,全血细胞减少,伴肝脾肿大
患者基本情况:
男性,14 岁。
主诉:
反复鼻出血2 月余。
现病史:
外院血常规提示全血细胞减少,2022 年8 月26 日就诊南方医院血液内科。
外院检查:
2022-05-16 外院骨髓:骨髓增生极度活跃,粒、红、巨三系增生均旺盛,巨核细胞
产板功能减低,血小板减少
2022-05-10 腹部彩超:肝脏增大,脾肝增大,脾肝高回声团。
2022-05-12CT:1.肝大、脾大2.肝左叶囊肿,肝左叶钙化灶
骨髓检查:
骨髓增生明显活跃,
粒:红=0.62:10
粒系:占32.0%增生相对减低,各阶段比值及形态大致正常。
红系:占52.0%增生明显活跃,以中晚幼红细胞增生为主,形态未见明显异常。成熟
红细胞胞体轻度大小不等,易见嗜多色性红细胞。
巨核系:环片一周见到巨核细胞469 个,分类100 个其中见到产板巨核细胞22 个;
血小板少见。
戈谢细胞占3.5%,此类细胞胞体大小悬殊,呈类圆形或不规则形,胞浆量丰富,淡蓝
色,呈洋葱皮样改变,可见交织成网状条纹样结构,胞核较小,单个或多个,呈不规
则形,可见扭曲折叠等现象,核染色质致密。
其它:淋巴细胞占12.5%部分形态不规则。
血象:无血片
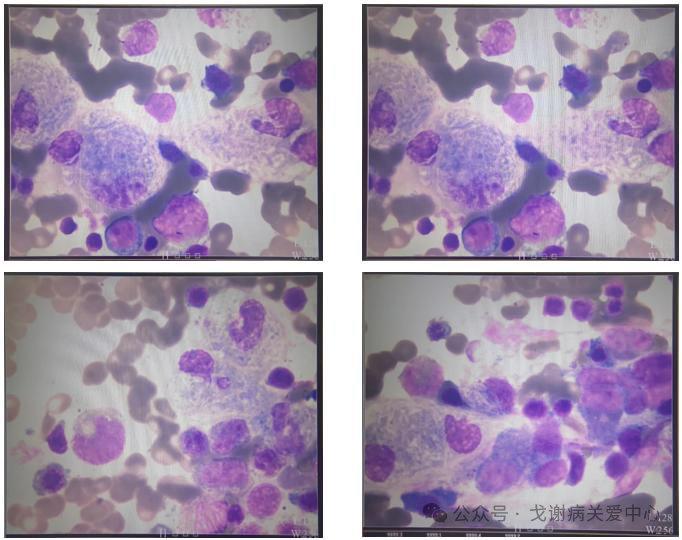
图骨髓穿刺结果(2022/8/31)
外院实验室酶学检查:
葡萄糖脑苷脂酶(GBA):0.44μmol/L /h(参考值1.26-22.23μmol/L/h)
诊断:
患者骨髓涂片见戈谢细胞,葡萄糖脑苷脂酶低于正常值,最终明确诊断为戈谢病Ⅰ型。
治疗:
患者从2023 年2 月起使用维拉苷酶α 8 支/次隔周治疗。治疗半年后血小板恢复至
150*109/L。
病例讨论
戈谢病(GD)是一种常染色体隐性遗传的溶酶体贮积症,其原因是机体葡萄糖脑苷脂
酶(GBA)基因发生变异,导致机体缺乏GBA 活性,导致其底物葡萄糖脑苷脂(也称
为葡萄糖神经酰胺)在肝、脾、骨骼、肺、脑等脏器的巨噬细胞溶酶体中贮积,从而
影响相应的组织和器官功能,继而出现相应的临床表现。
GBA 活性检测是GD 诊断的金标准。当患者出现不明原因的脾肿大,伴或不伴血小板
减少、贫血以及神经系统症状时,通过全血或干血纸片法(DBS)行GBA 活性检测,
若活性值低于参考值下限30%时,可诊断为GD[2]。不同分型患者的酶活性差异较大,
I 型患者通常表现出一定的残余酶活性,当GBA 活性值低于参考值下限但高于参考值
下限30%时,行GBA 基因检测可确诊。骨髓穿刺也是辅助检查之一,约30%左右的
患者骨髓检查能发现特征性细胞即“戈谢细胞”。该患者出现血细胞减少、肝脾肿大,
符合GD 的临床表现。实验室检查测得GBA 水平为0.44 μmol/L /h,低于参考值,
同时结合骨髓形态学检查发现特征性的“戈谢细胞”,最终确诊为GD。
在治疗方面,GD 的治疗方式包括非特异性治疗和特异性治疗。特异性治疗包括酶替代
治疗(ERT)、造血干细胞移植(HSCT)、底物减少疗法(SRT)、分子伴侣疗法和
基因疗法,其中ERT 是GD 的一线标准治疗方案,可通过特异性地补充患者体内缺乏
的酶,从而减少葡萄糖脑苷脂在体内的贮积。该患者目前正在接受维拉苷酶α这一新型
ERT 药物治疗,且取得较好的疗效,该药物于2021 年在我国获批上市,主要适用于
Ⅰ型GD 成人和4 岁以上的儿童患者[2]。根据我国共识[2],确诊戈谢病的儿童和青少年
一旦出现症状,均需考虑立即开始ERT,并建议长期使用。维拉苷酶α的建议起始剂量
为60U/Kg,治疗6 个月后,若内脏、血液学和生化指标无改善,需提高治疗剂量。
血小板计数在1~2 年内恢复至100×109/L 及以上时戈谢病患儿的治疗目标之一,该
患者经维拉苷酶α治疗半年后即达到该治疗目标,后续需继续密切观察症状及血液指标
变化,酌情调整治疗方案。
小结
本次我们报告了1 例儿童戈谢病患者的诊治经历。根据研究数据显示,GD 患病率仅
1/10 万[1],且发病年龄跨度很大,从婴儿期到成年都可能出现症状。儿童时期发病较
为常见[2],且临床表现较成人更为严重,甚至存在畸形和早期死亡的风险。我国最新
调研报告中显示1-20 岁的患者约占全部调研患者的63%[3],该患者发病年龄约为14
岁,属于青少年发病。
由于疾病的罕见性及症状的复杂多样性,导致临床对其关注度有限,且GD 的临床症
状与其他血液科的病相似,因此容易被误诊。研究表明,我国GD 患者的误诊时间平
均为3.74 年[4]。GD 的误诊和延迟诊断严重影响患者治疗,加重疾病负担,因此提高
其诊断及鉴别水平刻不容缓。
在治疗方面,ERT 是戈谢病的标准治疗方案,我国应用ERT 治疗GD 已有超过20 年
的临床使用经验,对于本例患者正在使用的维拉苷酶α,既往有研究显示,经维拉苷酶
α治疗后3 个月可观察到患者血红蛋白浓度和血小板计数等血液学指标改善,6 个月观
察到肝脾体积等内脏指标显著改善,在其长达7 年的扩展研究中,维拉苷酶α显示出持
久的疗效和长期达标[5,6]。对于经治患者而言,转换为维拉苷酶α后部分患者可见关键
临床参数进一步改善、Lyso-Gb1 水平继续下降,即“Booster-effect”(加强针效应)。
这些研究结果表明,维拉苷酶α在治疗戈谢病患者上具备独特优势。

蒋玲教授 南方医科大学南方医院
南方医科大学南方医院血液科副主任医师
以第一作者发表SCI论文5篇,核心期刊6篇;
获得国家自然科学基金1项;
获得广东省自然科学基金1项;
全国女医师协会白血病学专业组成员
广东省精准医学应用学会精准免疫治疗分会委员
广东省医师协会血液科医师分会白血病MDS专业组成员
广东省医师协会血液科医师分会红细胞疾病专业组成员
主要从事急性髓系白血病和MDS的基础研究,2019年赴美国MD.Anderson癌症研究中心从事博士后研究
参考文献:
[1] Nalysnyk L, Rotella P, Simeone J C, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature[J]. Hematology, 2017,22(2):65-73.
[2] 中华医学会儿科学分会内分泌遗传代谢学组, 中华医学会儿科学分会血液学组, 中华医学会医学遗传学分会, et al. 中国儿童戈谢病诊治专家共识(2021) [J]. 中华儿科杂志, 2021, 59(12): 7.
[3] 中国戈谢病患者诊疗状况及疾病负担调研报告2023,病痛挑战基金会,戈谢病病友会,艾社康健康咨询有限公司.
[4] 2019 中国戈谢病患者综合社会调查.中国罕见病联盟,北京协和医院,北京病痛挑战公益基金会.
[5] Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010 Jun 10;115(23):4651-6.
[6] Zimran A, Wang N, Ogg C,et al. Seven-year safety and efficacy with velaglucerase alfa for treatment-naïve adult patients with type 1 Gaucher disease. Am J Hematol. 2015 Jul;90(7):577-83.
审批号:C-ANPROM/CN/VPR/0070
审批日期:2024/1
仅供医疗卫生专业人士参考

 美摄影师为患病儿童拍超级英雄照
美摄影师为患病儿童拍超级英雄照 海南黎族文身即将消失
海南黎族文身即将消失 哪些人容易得慢阻肺
哪些人容易得慢阻肺 正常人如何预防脑出血
正常人如何预防脑出血 仁心在左 责任在右
仁心在左 责任在右 推己及人 以达极致
推己及人 以达极致 草莓吃腻了?这些吃法你肯定没试过!
草莓吃腻了?这些吃法你肯定没试过! 与癌共存:食管癌病人的自述
与癌共存:食管癌病人的自述 香味浓郁猪肉松
香味浓郁猪肉松 唐朝吉祥菜四喜丸子
唐朝吉祥菜四喜丸子 练起来!全身都要瘦!
练起来!全身都要瘦! 缓解痛经的九款药膳
缓解痛经的九款药膳