

6月初,有消息称恒瑞医药一处生产场地收到FDA 483表格,引发业内广泛关注,随后恒瑞方面做出回应。时隔一个月后,FDA官网公布了向“医药一哥”发布的警告信,从公开信息来看,此次收到的警告信与6月初公司收到的483表格为同一个关联事件,并没有显示FDA向恒瑞下发了进口禁令。恒瑞也向外界表示,目前公司该场地的产品出口未受影响,本次警告信对公司其他生产场地无影响,目前公司出口美国的产品也未受影响。
“一哥”收到警告信 称对公司业绩无重大影响
FDA警告信是给予违反美国《食品、药品和化妆品法案》的企业或个人的第一告知书。FDA检查官员对医药产品生产企业质量保证体系进行现场检查,并以483表的形式要求企业整改检查中出现的问题。若回复中未满足监管所有要求,即发出警告信。企业在收到此信的15个工作日内,需书面详细回复FDA,提供公司为纠正和预防此类问题再次发生所采取的相应措施。在企业完成整改后,FDA会开展针对公司整改措施的审评,在FDA认为公司已解决了警告信中提及的相关问题之后,将关闭警告信。
此前网上公开了一封记录时间为今年1月8日至1月16日的FDA 483表格,检查涉及恒瑞连云港地区一处制剂生产场地。近日,FDA官网显示,FDA对恒瑞位于江苏省连云港市经济技术开发区黄河路38号制剂生产场地GMP(药品生产质量管理规范)检查后出具了警告信,涉及的生产场地正是上述483表格所涉地址,说明了本次警告信是6月初483表的后续措施。
此次警告信中概述的内容主要为:质量控制部门未能履行其职责确保生产的制剂符合CGMP要求;设施设计不充分,未能在专门划定的、足够大的区域内进行生产操作,未能在无菌处理区域内设立独立或划定的区域或其他必要的控制系统来防止污染或混淆。
从目前公开信息来看,此次警告信所提出的缺陷与此前的483表所列问题基本一致,没有影响到药品质量安全,恒瑞医药也没有收到FDA进口禁令,说明FDA目前并未对产品质量有明确担忧。
据公开信息显示,恒瑞医药在全国有9个城市建有生产基地,仅连云港就有国际化制剂生产基地、新医药产业园、生物医药生产基地、原料药生产基地四个生产基地。据了解,此次警告信涉及场地出口美国的主要为一些仿制药制剂产品,目前共12个仿制药品种获FDA上市许可,但都不是公司主要产品。该场地2023年出口美国市场的收入仅占恒瑞当年营业收入的0.39%,2024年一季度出口美国市场的收入仅占恒瑞当期营业收入的0.47%。
恒瑞医药日前向外界回应,本次事件对公司其他生产场地无影响,预计不会对公司2024年业绩产生重大影响。
另据公开报道,今年7月2号,恒瑞医药连云港另一制剂生产场地的布比卡因脂质体注射液在美国成功获批上市。恒瑞作为复杂制剂全球首仿企业,说明其在药物研发、知识产权、生产质量等方面的综合实力较强。预计这个品种会增加恒瑞海外市场收益。
多次FDA检查顺利过关 这次是“一哥”突然松懈了?
从恒瑞医药官网可查询到,恒瑞首次向美国出口制剂产品是在2011年,抗肿瘤药伊立替康注射液获准在美国上市,公司成为第一家注射剂获准在美上市的民族制药企业。加上今年获批的他克莫司和布比卡因,至今恒瑞已有近20款仿制药进入美国市场,目前恒瑞产品已进入超40个国家,可谓出口经验丰富。
2022年年报显示:2019年,公司作为国内首家接受美国FDA总部PV系统视察的公司,在视察文件方面做到文件100%准备妥当、100%质量审阅,在系统流程方面做到FDA法规均有相应流程体现、流程执行均有文档可呈现,成为国内首家顺利通过美国FDA总部PV 系统视察、现场核查的公司。
由上可见其历史上在质量把控、合规管理等方面做得相当不错了。那么,以往FDA多次检查都顺利过关,这次突然收到警告信,是恒瑞方面突然松懈了,还是检查方面突然严格了?
在FDA网站上公布的警告信全文中,可以看到恒瑞医药针对所列的缺陷项目均已进行了整改。例如针对质量控制部门未能履行其职责确保生产的制剂符合CGMP要求的整改,FDA在警告信中提到:“在您的回复中,您表示将对文档管理软件进行风险重新评估,以便更好地控制相应规程。您创建了新的文件销毁的管理程序,修订了现有管理程序,并对您的员工进行了关于新的和修订的程序的培训。您的回复是不充分的。您的回复未包括对创建新记录以纠正错误的员工进行全面评估,也未确定相应‘错误’的根本原因或范围,以及对CGMP文件和数据可能产生影响。”
6月初有业内专家就483表分析称“预计美国FDA不会对恒瑞医药做出进一步的处罚或制裁”。从FDA的回应来看,没有否认恒瑞整改的方向,只是恒瑞这些改进计划的评估可能不够全面, 需要进一步完善,也可以视为FDA官方向企业提出了更高的标准和更严格的要求,可能也是恒瑞得到警告信的原因。
剑指中国企业?海外监管形势严峻
近二十年来,得益于低廉的成本效益和快速生产能力,中国一直是制药研究和制造服务的首选中心。然而,近年来的地缘政治紧张局势和“降低中国供应链风险”的呼声,让中国制造商面临打压。过去几年,美国参议院推出过两项新法案[1] [2],意在降低医药产品对外国的依赖,尤其是降低对中国等生产商的依赖。美国国内更是一直有加强对中国药企检查的呼声。
来看看下面这组数据,指向性已经相当明显。
根据FDA官方公布2019年-2024年合规行动措施(生物制品,医疗器械,人用药品)(https://datadashboard.fda.gov/ora/cd/complianceactions.htm)统计显示,FDA在2024年已经密集发布了134封警告信,呈现大幅增加的趋势。
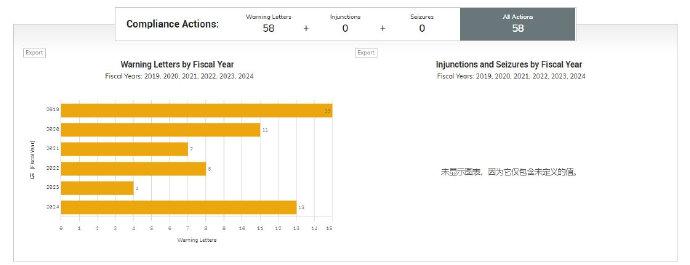
2019财年-2024财年 FDA向中国企业发布警告信数量汇总
从这张图可以看到,2024财年过去了9个月(2023年10月至2024年6月),中国企业警告信(13封)的数量已经远远超过了2023年全年。
与此同时,该合规行动措施显示,按照检查次数统计,美国、印度、中国分别为:10901、933、282,警告信出具比例分别为6.62%、7.07%、20.56%;美国的检查结果OAI(Official Action Indicated,强制采取整改,是FDA工厂审查的直接结果之一)比例5.72%,大致与警告信比例相同,而中国和印度OAI比例大致相同的情况下(中国、印度分别为9.75%、11.35%),FDA给中国企业的警告信比例却远远高于美国和印度。由此可以看出,FDA对中国和印度企业的监管和行动措施中对中国企业的容忍度更低。
创新药进入美国市场本就困难重重,但没想到如今仿制药挤进美国市场的这条路也更加艰难。
其实纵览全局进一步分析,美国的种种举动,似乎是对中国整个生物医药产业在美业务的发难。
今年5月,美国立法机构修改了《生物安全法案》,要求美国公司在2032年之前结束与药明康德等中国公司的合作,5月15日,美国众议院监督和问责委员会以压倒性票数批准了《生物安全法案》。虽然有消息称《生物安全法案》并没能纳入《国防授权法案》(NDAA)这个立法快速通道,但很多分析师认为该法案仍可能以其他方式通过。
事实上,美国众议院特设委员会在5月30日给FBI发信要求调研金斯瑞及其子公司的行为已经表明美国政府可能不止于仅打击中国CDMO。金斯瑞子公司传奇生物已是一家有上市产品的Biotech公司,且美国强生公司是合作伙伴,也被列入要求调查的范围。美国生物医药行业咨询公司BioCentury指出,“这封信明确将委员会的担忧从国家安全扩展到商业竞争力。”
更耐人寻味的是,6月6日,韩国、印度、美国、日本和欧盟正式发起成立了旨在合作建立有韧性供应链的“生物制药联盟”。此时,距离FDA公布恒瑞收到483表格仅过去了两天的时间。有媒体分析称,当下该联盟范围几乎已包含了国际上的主流市场。以美、日、欧盟为代表的全球创新药研发第一梯队,以三星生物等为代表的韩国CXO企业,再加上供应了美国60%以上过专利期仿制药的印度。“高、中、低端产品、原料药与供应链都聚齐了”。五方“合纵连横”,或许只为给中国生物医药产业的出海垒起一座不可逾越的城墙。
结合多种动向,恒瑞与FDA的此番博弈,也许已经不能仅作为个例来看了。
注释:
[1]https://baijiahao.baidu.com/s?id=1769379381912141793&wfr=spider&for=pc
[2]http://www.phirda.com/artilce_27259.html

 美摄影师为患病儿童拍超级英雄照
美摄影师为患病儿童拍超级英雄照 海南黎族文身即将消失
海南黎族文身即将消失 哪些人容易得慢阻肺
哪些人容易得慢阻肺 正常人如何预防脑出血
正常人如何预防脑出血 仁心在左 责任在右
仁心在左 责任在右 推己及人 以达极致
推己及人 以达极致 草莓吃腻了?这些吃法你肯定没试过!
草莓吃腻了?这些吃法你肯定没试过! 与癌共存:食管癌病人的自述
与癌共存:食管癌病人的自述 香味浓郁猪肉松
香味浓郁猪肉松 唐朝吉祥菜四喜丸子
唐朝吉祥菜四喜丸子 练起来!全身都要瘦!
练起来!全身都要瘦! 缓解痛经的九款药膳
缓解痛经的九款药膳